The things one can do when on sabbatical! For this week, it’s mainly attending the 13th Semantic Web Applications and tools for Health Care and Life Science (SWAT4HCLS) conference and even having some time to write a conference report again. (The last lost tagged with conference report was FOIS2018, at the end of my previous sabbatical.) The conference consisted of a tutorial day, two conference days with several keynotes and invited talks, paper presentations and poster sessions, and the last day a ‘hackathon’/unconference. This clearly has grown over the years from the early days of the event series (one day, workshop, life science).

It’s been a while since I looked in more detail into the life sciences and healthcare semantics-driven software ecosystems. The problems are largely the same, or more complex, with more technologies and standards to choose from that promise that this time it will be solved once and for all but where practitioners know it isn’t that easy. And lots of tooling for SARS-CoV-2 and COVID-19, of course. I’ll summarise and comment on a few presentations in the remainder of this post.
Keynotes
The first keynote speaker was Karin Verspoor from RMIT in Melbourne, Australia, who focussed her talk on their COVID-SEE tool [1], a Scientific Evidence Explorer for COVID-19 information that relies on advanced NLP and some semantics to help finding information, notably taking open questions where the sentence is analysed by PICO (population, intervention, comparator, outcome) or part thereof, and using UMLS and MetaMap to help find more connections. In contrast to a well-known domain with well-known terminology to formulate very specific queries over academic literature, that was (and still is) not so for COVID-19. Their “NLP+” approach helped to get better search results.
The second keynote was by Martina Summer-Kutmon from Maastricht University, the Netherlands, who focussed on metabolic pathways and computation and is involved in WikiPathways. With pretty pictures, like the COVID-19 Disease map that culminated from a lot of effort by many research communities with lots of online data resources [2]; see also the WikiPathways one for covid, where the work had commenced in February 2020 already. She also came to the idea that there’s a lot of semantics embedded in the varied pathway diagrams. They collected 64643 diagrams from the literature of the past 25 years, analysed them with ML, OCR, and manual curation, and managed to find gaps between information in those diagrams and the databases [3]. It reminded me of my own observations and work on that with DiDOn, on how to get information from such diagrams into an ontology automatically [4]. There’s clearly still lots more work to do, but substantive advances surely have been made over the past 10 years since I looked into it.
Then there were Mirjam van Reisen from Leiden UMC, the Netherlands, and Francisca Oladipo from the Federal University of Lokoja, Nigeria, who presented the VODAN-Africa project that tries to get Africa to buy into FAIR data, especially for COVID-19 health monitoring within this particular project, but also more generally to try to get Africans to share data fairly. Their software architecture with tooling is open source. Apart from, perhaps, South Africa, the disease burden picture for, and due to, COVID-19, is not at all clear in Africa, but ideally would be. Let me illustrate this: the world-wide trackers say there are some 3.5mln infections and 90000+ COVID-19 deaths in South Africa to date, and from far away, you might take this at face value. But we know from SA’s data at the SAMRC that deaths are about three times as much; that only about 10% of the COVID-19-positives are detected by the diagnostics tests—the rest doesn’t get tested [asymptomatic, the hassle, cost, etc.]; and that about 70-80% of the population already had it at least once (that amounts to about 45mln infected, not the 3.5mln recorded), among other things that have been pieced together from multiple credible sources. There are lots of issues with ‘sharing’ data for free with The North, but then not getting the know-how with algorithms and outcomes etc back (a key search term for that debate has become digital colonialism), so there’s some increased hesitancy. The VODAN project tries to contribute to addressing the underlying issues, starting with FAIR and the GDPR as basis.
The last keynote at the end of the conference was by Amit Shet, with the University of South Carolina, USA, whose talk focussed on how to get to augmented personalised health care systems, with as one of the cases being asthma. Big Data augmented with Smart Data, mainly, combining multiple techniques. Ontologies, knowledge graphs, sensor data, clinical data, machine learning, Bayesian networks, chatbots and so on—you name it, somewhere it’s used in the systems.
Papers
Reporting on the papers isn’t as easy and reliable as it used to be. Once upon a time, the papers were available online beforehand, so I could come prepared. Now it was a case of ‘rock up and listen’ and there’s no access to the papers yet to look up more details to check my notes and pad them. I’m assuming the papers will be online accessible soon (CEUR-WS again presumably). So, aside from our own paper, described further below, all of the following is based on notes, presentation screenshots, and any Q&A on Discord.
Ruduan Plug elaborated on the FAIR & GDPR and querying over integrated data within that above-mentioned VODAN-Africa project [5]. He also noted that South Africa’s PoPIA is stricter than the GDPR. I’m suspecting that is due to the cross-border restrictions on the flow of data that the GDPR won’t have. (PoPIA is based on the GDPR principles, btw).
Deepak Sharma talked about FHIR with RDF and JSON-LD and ShEx and validation, which also related to the tutorial from the preceding day. The threesome Mercedes Arguello-Casteleiro, Chloe Henson, and Nava Maroto presented a comparison of MetaMap vs BERT in the context of covid [6], which I have to leave here with a cliff-hanger, because I didn’t manage to make a note of which one won because I had to go to a meeting that we were already starting later because of my conference attendance. My bet would be on the semantics (those deep learning models probably need more reliable data than there is available to date).
Besides papers related to scientific research into all things covid, another recurring topic was FAIR data—whether it’s findable, accessible, interoperable, and reusable. Fuqi Xu and collaborators assessed 11 features for FAIR vocabularies in practice, and how to use them properly. Some noteworthy observations were that comparing a FAIR level makes more sense before-and-after changing a single resource compared to pitting different vocabularies against each other, “FAIR enough” can be enough (cf. demanding 100% compliance) [7], and a FAIR vocabulary does not imply that it is also a good quality vocabulary. Arriving at the topic of quality, César Bernabé presented an analysis on the use of foundational ontologies in bioinformatics by means of a systematic literature mapping. It showed that they’re used in a range of activities of ontology engineering, there’s not enough empirical analysis of the pros and cons of using one, and, for the numbers game: 33 of the ontologies described in the selected literature used BFO, 16 DOLCE, 7 GFO, and 1 SUMO [8]. What to do next with these insights remains to be seen.
Last, but not least—to try to keep the blog post at a sort of just about readable length—our paper, among the 15 that were accepted. Frances Gillis-Webber, a PhD student I supervise, did most of the work surveying OWL Ontologies in BioPortal on whether, and if so how, they take into account the notion of multilingualism in some way. TL;DR: they barely do [9]. Even when they do, it’s just with labels rather than any of the language models, be they the ontolex-lemon from the W3C community group or another, and if so, mainly French and German.

Does it matter? It depends on what your aims are. We use mainly the motivation of ontology verbalisation and electronic health records with SNOMED CT and patient discharge note generation, which ideally also would happen for ‘non-English’. Another use case scenario, indicated by one of the participants, Marco Roos, was that the bio-ontologies—not just health care ones—could use it as well, especially in the case of rare diseases, where the patients are more involved and up-to-date with the science, and thus where science communication plays a larger role. One could argue the same way for the science about SARS-CoV-2 and COVID-19, and thus that also the related bio-ontologies can do with coordinated multilingualism so that it may assist in better communication with the public. There are lots of opportunities for follow-up work here as well.
Other
There were also posters where we could hang out in gathertown, and more data and ontologies for a range of topics, such as protein sequences, patient data, pharmacovigilance, food and agriculture, bioschemas, and more covid stuff (like Wikidata on COVID-19, to name yet one more such resource). Put differently: the science can’t do without the semantic-driven tools, from sharing data, to searching data, to integrating data, and analysis to develop the theory figuring out all its workings.
The conference was supposed to be mainly in person, but then on 18 Dec, the Dutch government threw a curveball and imposed a relatively hard lockdown prohibiting all in-person events effective until, would you believe, 14 Jan—one day after the end of the event. This caused extra work with last-minute changes to the local organisation, but in the end it all worked out online. Hereby thanks to the organising committee to make it work under the difficult circumstances!
References
[1] Verspoor K. et al. Brief Description of COVID-SEE: The Scientific Evidence Explorer for COVID-19 Related Research. In: Hiemstra D., Moens MF., Mothe J., Perego R., Potthast M., Sebastiani F. (eds). Advances in Information Retrieval. ECIR 2021. Springer LNCS, vol 12657, 559-564.
[2] Ostaszewski M. et al. COVID19 Disease Map, a computational knowledge repository of virus–host interaction mechanisms. Molecular Systems Biology, 2021, 17:e10387.
[3] Hanspers, K., Riutta, A., Summer-Kutmon, M. et al. Pathway information extracted from 25 years of pathway figures. Genome Biology, 2020, 21,273.
[4] Keet, C.M. Transforming semi-structured life science diagrams into meaningful domain ontologies with DiDOn. Journal of Biomedical Informatics, 2012, 45(3): 482-494. DOI: dx.doi.org/10.1016/j.jbi.2012.01.004.
[5] Ruduan Plug, Yan Liang, Mariam Basajja, Aliya Aktau, Putu Jati, Samson Amare, Getu Taye, Mouhamad Mpezamihigo, Francisca Oladipo and Mirjam van Reisen: FAIR and GDPR Compliant Population Health Data Generation, Processing and Analytics. SWAT4HCLS 2022. online/Leiden, the Netherlands, 10-13 January 2022.
[6] Mercedes Arguello-Casteleiro, Chloe Henson, Nava Maroto, Saihong Li, Julio Des-Diz, Maria Jesus Fernandez-Prieto, Simon Peters, Timothy Furmston, Carlos Sevillano-Torrado, Diego Maseda-Fernandez, Manoj Kulshrestha, John Keane, Robert Stevens and Chris Wroe, MetaMap versus BERT models with explainable active learning: ontology-based experiments with prior knowledge for COVID-19. SWAT4HCLS 2022. online/Leiden, the Netherlands, 10-13 January 2022.
[7] Fuqi Xu, Nick Juty, Carole Goble, Simon Jupp, Helen Parkinson and Mélanie Courtot, Features of a FAIR vocabulary. SWAT4HCLS 2022. online/Leiden, the Netherlands, 10-13 January 2022.
[8] César Bernabé, Núria Queralt-Rosinach, Vitor Souza, Luiz Santos, Annika Jacobsen, Barend Mons and Marco Roos, The use of Foundational Ontologies in Bioinformatics. SWAT4HCLS 2022. online/Leiden, the Netherlands, 10-13 January 2022.
[9] Frances Gillis-Webber and C. Maria Keet, A Survey of Multilingual OWL Ontologies in BioPortal. SWAT4HCLS 2022. online/Leiden, the Netherlands, 10-13 January 2022.



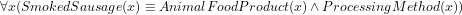 (which is ontologically incorrect because a smoked sausage is not way of processing) or, if done with a ‘cross-product’ and a new relation (
(which is ontologically incorrect because a smoked sausage is not way of processing) or, if done with a ‘cross-product’ and a new relation ( ), then the resulting computation will have something alike
), then the resulting computation will have something alike  instead of having declared directly in the ontology proper, say,
instead of having declared directly in the ontology proper, say, 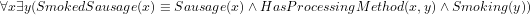 . The latter option has the advantages that it makes it easier to add, say, Fermented smoked sausage or Cooked smoked sausage as a sausage that has the two properties of being [fermented/cooked] and being smoked, and that one can avail of automated reasoners to classify the taxonomy. Either way, the details are being worked on. The ontology language and the choice for one or the other—whichever it may be—ought not to get in the way of developing an ontology, but, generally, it does so both regarding underlying commitment that the language adheres to and any implicit or explicit workaround in the modelling stage that to some extent make up for a language’s limitations.
. The latter option has the advantages that it makes it easier to add, say, Fermented smoked sausage or Cooked smoked sausage as a sausage that has the two properties of being [fermented/cooked] and being smoked, and that one can avail of automated reasoners to classify the taxonomy. Either way, the details are being worked on. The ontology language and the choice for one or the other—whichever it may be—ought not to get in the way of developing an ontology, but, generally, it does so both regarding underlying commitment that the language adheres to and any implicit or explicit workaround in the modelling stage that to some extent make up for a language’s limitations.








